- 大分類
-
- 血小板
- 小分類
-
- 疾患
血小板無力症 Glanzmann thrombasthenia
解説
1.病態・病因
先天性血小板機能異常症の一つ。1918年にスイスのGlanzmann博士により血小板数が正常で血餅退縮能に異常を伴う出血性疾患群として初めて報告された。本疾患では、血小板凝集に必要なフィブリノゲンおよびフォン・ヴィレブランド因子(VWF)の受容体であるGPIIb-IIIaが欠損(あるいは機能障害)している。
GPIIb/IIIa(インテグリンαIIb/β3)は接着タンパク受容体ファミリーであるインテグリンに属し、GPIIb(αIIb)とGPIIIa(β3)が Caイオン依存性に1:1の比率にて複合体を形成し、血小板あたり約80,000分子と最も豊富に存在する接着タンパク受容体である。巨核球/血小板系に特異的に発現している。
2.疫学
約50万人に1人
3.診断
1)常染色体劣性遺伝形式である。両親は出血傾向を示さない。本疾患はGPIIb/IIIaの量的異常と質的異常(variant型)に分類される。
2)幼少時より紫斑、点状出血などの皮膚症状、鼻血や消化管出血などの粘膜出血が見られ、大人になるに従って症状が軽くなる傾向にある。血小板数は正常であり、出血時間は著明に延長する(15分以上)。血小板凝集能検査において、リストセチン凝集を除く、アデノシン二リン酸(ADP)、コラーゲン、エピネフリンなどで惹起されるフィブリノゲン依存性の血小板凝集がすべて欠如する(図)。血餅退縮能は低下あるいは欠如している。
3)遺伝子異常はGPIIbあるいはGPIIIa遺伝子のどちらか一方のみに存在。GPIIbとGPIIIaは複合体を形成するため、どちらかの一方の異常により複合体として発現されない結果、GPIIbとGPIIIaの両者が欠損する。
4.治療
治療法としては、重篤な出血や手術の時などには血小板輸血を行う。遺伝子組換え活性型第VII因子(FVIIa)製剤(ノボセブン®)が血小板輸血不応の血小板無力症に対して適用となっている。
図表
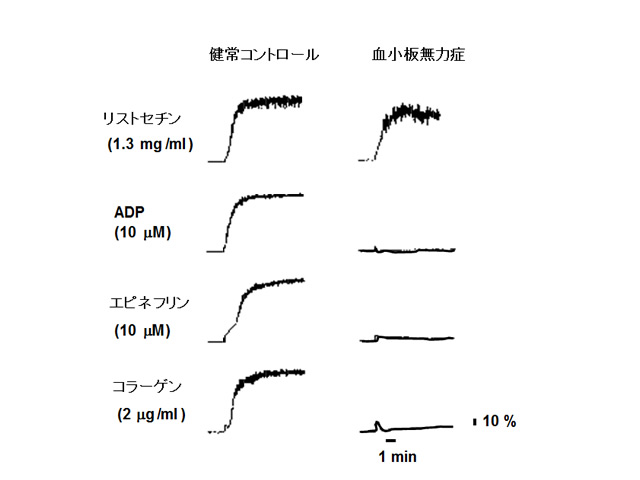
図 血小板無力症患者の血小板凝集曲線
リストセチン凝集を除く、ADP凝集、コラーゲン凝集、エピネフリン凝集がすべて欠如する。(血栓止血学会誌16:171-178, 2005より引用改変)
引用文献
1) 冨山佳昭:血小板無力症,血栓止血誌 16:171-178,2005.